What Mad Pursuit (17 page)
Authors: Francis Crick

A good model in biology, then, not only should address the problem in hand but if at all possible should serve to unite evidence from several different approaches so that various sorts of tests can be made of it. This may not always be possible to do straight away—the theory of natural selection could not immediately be tested at the cellular and the molecular level—but a theory will always command more attention if it is supported by unexpected evidence, particularly evidence of a different
kind.
The Missing Messenger
T
HE NEXT EPISODE I want to touch on concerns what we now call messenger RNA. The double-helical structure of DNA had given us a theoretical framework that was invaluable as a guide to research, since it not only tied together approaches that at first sight seemed to have no connection with each other, but it suggested radically new experiments that could not have been conceived without the DNA model as a guide. Unfortunately, our thinking contained one major error. It was uncertain at that time whether any protein synthesis took place in the nucleus of the cell (where most of the DNA was), but everything suggested that the majority of it took place in the cytoplasm. In some way the sequence information in the nuclear DNA had to be made available outside the nucleus, in the cytoplasm. The obvious idea, which predated the DNA model, was that this messenger was RNA. This was the basis of the slogan coined by Jim Watson: “DNA makes RNA makes protein.”
It was known that cells very active in protein synthesis had more RNA in their cytoplasm than cells that were less active. By the late 1950s it had been shown that most of their RNA was in small particles, now named ribosomes, that consisted of RNA molecules plus a mixture of proteins. What more natural than to assume that each ribosome synthesized just one protein and that its RNA was the postulated messenger RNA? We assumed that each active gene produced a (single-stranded) RNA copy of itself, that this was packaged in the nucleus with a set of proteins to help it do its job and then exported to the cytoplasm where it directed the synthesis of the particular polypeptide chain coded for by this RNA. Each ribosome, working in concert with the transfer-RNA molecules (see
appendix A
), would in some way embody the details of the genetic code (surmised, but not yet discovered) so that the four-letter language of the RNA could be translated into the twenty-letter language of the proteins.
About this time, Sydney Brenner and I discussed at some length how we could prove this idea by isolating a single ribosome, supplying it with all the necessary precursors, and then showing that it produced just one type of protein. Fortunately the problem seemed hopelessly difficult, as the techniques then available were not sensitive enough. We might have wasted much time and effort in difficult experiments that, unbeknown to us, were bound to fail.
Since ribosomes were obviously important structures, there was much experimental work on them. The techniques used were often new ones and thus open to suspicion, and the results were seldom clear-cut. Nevertheless, a whole series of awkward “facts” started to press for attention. The ribosomal RNA in a growing bacterial cell hardly seemed to turn over at all and was therefore described as “an inert metabolic product.” RNA molecules in the ribosomes would have been expected to vary in length, since one protein is often a very different length from another. Yet the experiments indicated that the ribosomal RNA came in just two fixed sizes. The base composition of DNA in different species of bacteria varied over a wide range. Their messenger RNA might be expected to vary in the same way, yet the composition of the postulated messenger, the ribosomal RNA, varied only a little in these very different species. We could invent ad hoc reasons to explain away all these weak facts, but they made us very uncomfortable. Sydney and I spent many long hours going over the evidence, trying to spot what was wrong.
As it turned out, clarification came from a quite different source. The group of workers at the Institut Pasteur in Paris had carried out an experiment known as the PaJaMo experiment, because the authors were Arthur Pardee (a visiting American), Jacob, and Monod. Monod’s interest was mainly in the formation of induced enzymes and in particular in the enzyme β-galactosidase The cell switched on the synthesis of this enzyme if the sugar galactose was supplied to it instead of the more customary glucose. Jacob’s main concern was how genetic information was passed between cells during mating. He and Eli Wollman had done the famous blender experiment on bacteria in which the “male” and “female” cells had been allowed to join together and then, after a chosen time, had been separated by putting them in a Waring blender, an example of molecular coitus interruptus. Fortunately the process of mating is a prolonged one (it can last up to two hours, equivalent to several normal lifetimes for a rapidly growing cell), which makes it easier to study. They had shown that the genes were transferred in a linear fashion over this period, in a fixed order, so that interrupting the process had little effect on the earlier genes but prevented the transfer of the later ones. This turned out to be the key discovery in bacterial genetics, clearing up a whole series of complications and difficulties that had accumulated over the years.
From our point of view the significant aspect of this process was that a particular gene, such” as the gene for β-galactosidase, would be introduced into the cell at a known time. It was then possible to see how the synthesis of this new protein changed with time, after the gene was introduced into the cell.
The result was surprising. We would have expected that the new gene started fairly soon to produce its own ribosomes, that these would accumulate slowly, and that as more and more ribosomes came into operation protein synthesis would steadily accelerate. The PaJaMo experiment showed something quite different. Very shortly after the gene was introduced the synthesis of β-galactosidase started up at a fairly fast rate and stayed that way.
Naturally we were reluctant to believe this experiment. Jacques Monod had first told us about it when he visited Cambridge, but at that stage the results were preliminary. Sydney and I worried about it during the following months. I tried to devise some way out, but my attempts seemed very forced.
A little later François Jacob came to Cambridge and on Good Friday, 1960, when the laboratory was closed, a small group of us assembled in a room in the Gibbs Building of King’s College, of which Sydney was a Fellow. Horace Judson has given a much fuller account of the whole episode. Here I will only touch on the main points.
I started by cross-examining François about the PaJaMo experiment, since there were several possible loopholes in the original paper. François detailed to us how the experiments had been improved. He also reported a very recent experiment by Pardee and Monica Riley in Berkeley. Slowly we realized that we would have to accept the results as correct. Exactly what happened then is obscure, since it has been obliterated in what follows, but the train of thought can easily be reconstructed. What the PaJaMo type of experiment showed was that the ribosomal RNA could not be the message. All the previous difficulties had prepared us for this idea, but we had not been able to take the necessary next step, which was: Where, then, is the message? At this point Sydney Brenner let out a loud yelp—he had seen the answer. (So had I, for that matter, though nobody else had.) One of the peripheral problems of this confused subject had been a minor species of RNA that occurred in
E. coli
shortly after it had been infected by bacteriophage T4.
(E. colt
, a bacterium that lives in our gut, is much used in the laboratory.) Some years earlier, in 1956, two workers, Elliot Volkin and Lazarus Astrachan, had shown that a new species of RNA was synthesized that had an unusual base composition, since it mirrored the base composition of infecting phage and not that of the
E. coli
host, which happened to be very different. They had at first thought that this might be the precursor of the phage DNA, which the infected cell was compelled to synthesize in large quantities, but further careful work on their part had shown that this hypothesis was incorrect. Their result had hung in midair, surprising but unexplained.
The problem was then: If messenger RNA was a different species of RNA from ribosomal RNA, then why had we not seen it? What Sydney had seen was that the Volkin-Astrachan RNA
was
the messenger RNA for the phage-infected cell. Once this key insight had been obtained, the rest followed almost automatically. If there was a separate messenger RNA, then clearly a ribosome did not need to contain the sequence information. It was just an inert reading head. Instead of one ribosome being tied to the synthesis of just one protein, it could travel along one message, synthesizing one protein, and then go onto a further messenger RNA, where it would synthesize a different protein. The PaJaMo results were easily explained by assuming that the messenger RNA was used only a few times before being destroyed. (We thought at first that it might be used only once but soon saw that this was unnecessarily restrictstive.) This explained the linear increase of protein with time, since the messenger RNA for /3-galactosidase soon reached an equilibrium concentration in which messenger synthesis was balanced by messenger degradation. This sounded wasteful but it allowed the cell to adjust rapidly to changes in its environment.
That evening I held a party at the Golden Helix. We often had parties (the molecular biologists’ parties were considered to be the liveliest in Cambridge), but this one was different. Half the guests, such as the virologist Roy Markham, who had not been at the morning meeting, were just having a good time. The other half, in small groups, were earnestly discussing the new idea, seeing how it easily explained puzzling data and actively planning radically new key experiments to put the hypothesis to the test. Some of these were done later by Sydney, on a visit to Cal Tech, with François and Matt Meselson.
It is difficult to convey two things. One is the sudden flash of enlightenment when the idea was first glimpsed. It was so memorable that I can recall just where Sydney, François, and I were sitting in the room when it happened. The other is the way it cleared away so many of our difficulties. Just a single wrong assumption (that the ribosomal RNA was the messenger RNA) had completely messed up our thinking, so that it appeared as if we were wandering in a dense fog. I woke up that morning with only a set of confused ideas about the overall control of protein synthesis. When I went to bed all our difficulties had resolved and the shining answers stood clearly before us. Of course, it would take months and years of work to establish these new ideas, but we no longer felt lost in the jungle. We could survey the open plain and clearly see the mountains in the distance.
The new ideas opened the way for some of the key experiments used to crack the genetic code, since one could now conceive
adding
special messengers to ribosomes (either natural messengers or synthetic ones), an idea that before had no meaning.
Naturally you may be compelled to ask: Why had we not seen it before? In a certain sense we had, but because nothing seemed to support it we had not recognized it as important. It required us to accept, first, that the RNA we
did
see in the cytoplasm was
not
the messenger RNA and therefore had some other function. Exactly what that function is, is not clear even to this day, though we can make educated guesses. It also required us to hypothesize a keyspecies of RNA
that had never been seen.
I wish I had been bold enough to take this step, but my natural caution must have prevented me. The irony was, of course, it had been seen in one particular case (the phage-infected cell) but we had not recognized it till that fateful Good Friday morning. Of course, messenger RNA was bound to be discovered eventually, but there is little doubt in my mind that this revelation speeded up the process considerably. After that, it might be said, the experiments wrote themselves. Nothing remained but the hard work: a happy state of affairs.
Triplets
A
LTHOUGH SYDNEY AND I clearly realized that the genetic code was a biochemical problem, we still had hopes that genetic methods could contribute to the solution, especially as genetic methods, using the right material, can be very fast, whereas biochemical methods are often rather slower. Seymour Benzer had used genetic methods to demonstrate that the genetic material was almost certainly one-dimensional. The question had been inspired by the DNA double helix but the method used was entirely original.
In order to map a gene very finely, it is necessary to pick up rather rare individuals. The nearer two mutants are in a gene, the rarer will be the act of genetic recombination between them. Benzer had chosen a system with two advantages. The genes in question were in the bacteriophage T4, a virus that attacked and killed the cells of
E. coli.
The virus grows fast and recombines at a high rate. He had chosen the gene called r
II
—actually a pair of genes next to each other—because it had a remarkable technical advantage. By using appropriate strains of the host cell, it was possible to pick up one virus haying a wild-type gene even if mixed with millions of viruses with the mutant version. Thus very rare recombinant genes could be detected, so rare that Benzer calculated that even adjacent base pairs on the DNA could be separated. Unfortunately there was no corresponding method for picking up a mutant among a vast excess of wild type, but, on the appropriate host the plaque—the little colony formed by the growth of one bacterium in the lawn of
E. coli
in the petri dish—looked different and was easy to recognize. A single mutant plaque in a petri dish with several hundred wild-type plaques could be spotted fairly easily.
The conventional way to map would have been to pick a series of distinct mutants and then find the recombinant distance between any pair of them. More elaborate methods using three mutants were also possible, but all these involved the counting of hundreds and thousands of plaques, which was very laborious.
Benzer, always one to avoid unnecessary work, thought of a better method. As well as point mutations, he found that some of his mutants appeared to be deletions. They mapped as lines on his genetic map, since they appeared to overlap at least two of the point mutations. He was thus able to collect a whole series of deletions. If two deletions overlapped then genetic recombination could never give back the intact wild type, since the overlapping portion was in neither parent and thus could not be regained. On the other hand, if the two deletions did
not
overlap, then an appropriate recombination event could restore the wild type.
An analogy may help to make this clearer. Imagine two defective copies of a book, one with pages 100 to 120 missing and the other with pages 200 to 215 missing. Clearly from these two copies, each with a single, contiguous deletion, we could recover the text of the complete book. However, if the second book, instead of having pages 200 to 215 missing, lacked pages 110 to 125, there would then be no way of recovering pages 110 to 120, since they would be missing in both copies.
To make the analogy closer we have to extend it a little. Imagine that the book contained very detailed instructions for making a complicated instrument. Assume also that if any one page were missing, then either no instrument would be made or, if it were, it would be a dud one. Finally, assume that we had millions of copies of each of the defective books. The rule then was: Select one copy of each type of book. Take the first
n
pages from one book and the remaining pages from the other. See if this new hybrid book will produce an instrument that works. Do this a million times, selecting the cross-over page (page
n
) at random each time. If occasionally a good instrument was produced, the two deletions did
not
overlap. If a good instrument was
never
produced, then the deletions probably overlapped.
This may seem an elaborate way of doing it, but since we could not look inside the phage this was the only method we had. All Benzer had to do, then, was to mate the two viruses together, by simultaneously infecting a culture of
E. coli
with them. After they had grown and recombined inside the bacteria, the viruses could be plated on a petri dish having the special host strain. If the deletions did not overlap, then there would be some recombinant plaques on the plate. If they
did
overlap, there would be none. There was no need for laborious counting. All that was needed was a simple yes or no answer.
Benzer argued that if the gene was two-dimensional then eventually he should find a special pattern with four deletions. Deletion
α
would overlap
B
and
C
, as would
D;
but deletions
B
and
C
would not overlap nor would
α
and
D.
(See
figure 12.1.
) It is easy to see that this cannot happen if the gene is one-dimensional. Benzer picked up hundreds of deletions and crossed them all against one another in pairs. The situation shown in the figure never arose. Therefore, he concluded, the gene is probably one-dimensional. His results also allowed him to put all his deletions in order, so he would see roughly where each one was in the map of the gene.
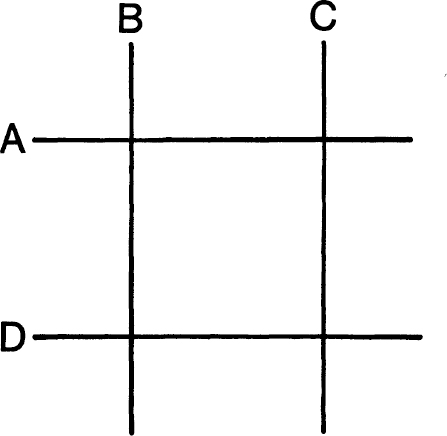
To show that in
two
dimensions we can have A overlapping both B and C, and D also overlapping B and C, without A overlapping D or B overlapping C. This is impossible if A, B, C, and D are segments of a (one-dimensional) straight line. Since Benzer never found this pattern of overlapping among his many deletions, he concluded, correctly, that the gene he was studying was one-dimensional. This was compatible with it being made of DNA.
For a variety of reasons, our group had chosen the same system to work on. Our main interest was in the different type of mutants produced by different chemicals and also in the reverse mutations these chemicals produced. Mutants appeared to fall broadly into two classes. Most chemicals produced mutants of the first class. However, the mutants produced by chemicals of the acridine type fell into the other. Each class was most easily reverted by the type of mutagen that produced it. Ernst Freese had suggested that one class corresponded to transitions (purine to purine, or pyrimidine to pyrimidine—see
appendix A
) while the other corresponded to transversions, as they were called (purine to pyrimidine, or vice versa). We had come up with another idea. Some mutants were leaky—that is, they showed the gene was active to some extent, though of course not fully active—whereas others were nonleaky—that is, had essentially no activity. We noticed that mutants produced by proflavin (a typical acridine) were almost always non-leaky. This led us to suggest that the proflavin mutants were tiny deletions or additions to the base sequence, whereas all the other class of mutants were base substitutions of one sort or another. However, we lacked further evidence to confirm this idea.
Meanwhile I had come up with a quite different idea. Pondering over how an RNA molecule could act as a message, I wondered if it could fold back on itself, thus forming a loose double-helical structure. The idea was that some bases could pair whereas others, which did not match according to the pairing rules, would loop out. The “code” would then depend either on the looped-out bases or the paired ones, or some more elaborate combination of these two possibilities. The idea was really rather vague, but it made one important prediction. A mutant at one end of the message might, in theory, be capable of being compensated in its effect by another, toward the other end, that paired with it. Thus some mutants should have distant “suppressors,” as they are called, within the same gene.
I rather liked this idea, but nobody else thought very much of it. Up to that time I had not done any phage genetics myself, being content to look at the experimental results of my colleagues. Since nobody seemed keen to test my idea, I decided to test it myself. It was not difficult to learn how to do phage genetics, especially with expert help at hand. In spite of this I made some elementary mistakes, fortunately soon corrected. The experiments also taught me how superficial my knowledge was, even though I had taken part in numerous discussions about this very system. There is nothing like actively doing experiments to make one realize all the ins and outs of a technique. It also helps to fix the details in one’s head, especially as reading the “experimental methods” section of most scientific papers is more boring than almost anything I know.
Naturally I chose the r
II
genes for the experiment, concentrating on the second one, the so-called B cistron. (Cistron was Benzer’s fancy name for a gene, as defined by the so-called cis-trans test.) I selected a mutant from our stock, tried to find a revertant—one more like the wild type—and then looked to see if this reversion was due to a second mutation somewhere else in the same gene. If I couldn’t find it I went on and tried again with another mutant.
At first I couldn’t find any suppressors. Presumably the change that reverted the mutant to wild type was at, or very close, to the original change, too close for me to pick it up. Leslie Orgel came over to coffee one day. As he looked over my shoulder I told him what I was doing and that so far I had no results. He went off to join the others while I quickly scored the remaining plates. To my delight I found I had a candidate suppressor.
Before long I had three mutants with suppressors, fortunately spaced out along the map. I isolated the suppressors and proceeded to map them. My theory was immediately refuted. Instead of each suppressor mapping at the predicted place, some distance away on the map, each had its suppressor quite near it. The suppressor effect must be due to some other reason.
Unbeknown to me, other people had also noted that a mutant in r
II
could have a suppressor in the same gene. Perhaps the most striking example occurred at Cal Tech. Dick Feynman, the theoretical physicist, had become sufficiently interested in these genetic problems that he had decided to do some experiments himself. He stumbled upon an example of an internal suppressor. Not knowing what it might imply, he asked his mentor, Max Delbrück. Max suggested that the original mutant had produced a changed amino acid and that the second mutant had changed another amino acid, elsewhere in the protein, which somehow compensated for the first change. It was easy to see that this might happen, but one would not expect it to be too common.
I was certainly aware of this possibility but I was not happy with it, partly because I had a very detailed knowledge of what little was then known about protein structure. I decided to try to see how many different suppressors a particular mutant might have. I had to select one of my three to study further and, sensibly, I chose the one whose suppressor was the least close to the parent mutant, hoping this would give me more elbow room. I also noted that two of my three mutants had been produced by proflavin. Even though this was hardly statistically significant, by any test, it seemed suggestive to me.
By now I was a little more experienced, so the experiments went fairly quickly. Phage genetics has the advantage that experiments are rather fast, once everything is set up. It does not take long to carry out a hundred crosses, since the manipulations are easy and an actual cross takes only about twenty minutes, this being the time for the phage to infect the bacterium, to multiply inside it (exchanging genetic material in the process), and to burst open, thus killing the cell. The results of the cross must then be plated out on petri dishes, to which a thin film of bacteria has been added. Then the dishes have to be incubated, to produce a lawn of bacteria. Where a single phage has landed and infected a cell, a colony of phage will grow, killing the local bacteria as it does so, forming a clear little hole (called a plaque) in the lawn of growing bacteria on the surface of the plate. This process takes a few hours, so one has a brief respite while it is going on. Then the petri dishes have to be taken from the 37 ° C incubator and examined to see whether they have plaques or not and, if so, of what type. Interesting plaques are then “picked"—that is, a few phage are picked up with a little piece of paper or a toothpick; grown further; and the process repeated a second time to make sure the phage stock is a pure one. If one works reasonably hard it is possible to complete one extra set of crosses in one day, and prepare for a new set the next day.
As the experiments got more interesting I found that, with careful planning, I could get through
two
successive sets of crosses in one day. This involved starting promptly in the morning, going home for lunch, more experiments in the afternoon, home for dinner, and a final set after dinner. Fortunately Odile and I then lived within a few minutes of the laboratory, an easy walk through the center of historic Cambridge, so I did not find the work unpleasant. In fact, Odile has told me she had never seen me so cheerful as during the period when I did experiments all the time, but this may have been partly because, for weeks on end, all the experiments seemed to work perfectly.