Ancient DNA: Methods and Protocols (32 page)
Read Ancient DNA: Methods and Protocols Online
Authors: Beth Shapiro

Volume ( m L)
Final concentration
Reagent
per sample
in reaction
Water (add 20 m L)
4.95
10× buffer (10×)
2
1×
25 mM MgCL
1.6
2 mM
2
dNTPs (25 mM each)
0.2
0.25 mM each
ext_ primer_F (10 m M)
0.5
0.25 m M
ext_ primer_R (10 m M)
0.5
0.25 m M
Amplitaq Gold (5 U/ m L)
0.25
1.25 U
Template
10
AMPure XP DNA purifi cation kit. Further evaluate the success of the barcoding procedure by verifying the expected size-shift from your template DNA to the increased size of the fi nal library (sum of template DNA and the length of both adapters) (see Note 12).
2. Following quantifi cation, the double-stranded libraries can be pooled in equimolar ratios and submitted to the standard 454
sequencing pipeline.
4. Notes
1. A low concentration of Tween 20 in the elution buffer can prevent DNA molecules from sticking to the tube walls. As a result, less DNA is lost when transferring DNA between reaction tubes. Tween-20 also improves handling of SPRI and Streptavidin beads.
2. Alternatively to this protocol, the 5 ¢ and 3 ¢ adapters can also be adjusted to 100 m M each and mixed to produce a ready-to-use adapter mix. However, if both adapters carry barcodes, pre-mixing the adapters will limit the number of possible barcode combinations.
3. Prolonged incubation may cause recessed instead of blunt ends and reduce ligation effi ciency.
4. It is essential to combine DNA and adapters fi rst prior to adding the master mix containing the ligase. If the adapters are 19 Generating Barcoded Libraries for Multiplex High-Throughput Sequencing 169
added to the master mix containing the ligase, large amounts of adapter dimers may form. Adding the master mix to the
DNA before adding the adapters may increase the number of
chimeric, concatenated target molecules.
5. To reduce the amount of unincorporated adapters, two PE
washes are recommended at this step.
6. As very low copy number aDNA extracts are diffi cult to quantify before adapter ligation, the exact amount of adapters needed for the ligation cannot easily be calculated. Therefore, an excess of adapters is usually used. However, excess adapters and adapter dimers can signifi cantly reduce the number of reads on target (reads that map to the target regions) produced by the NGS instrument. It is therefore essential to reduce the amount of dimers and unincorporated adapters in the library as much as possible by performing the washing procedure
described under Subheading 3.2.4
.
7. Siliconized tubes prevent DNA from sticking to tube walls.
Thus less DNA is lost through repeated freeze/thaw cycles.
8. In case of using fully amplifi ed PCR products (>10 ng/ m L) as a template, 15 m L of a 1:10 dilution of the purifi ed PCR product will be suffi cient.
9. To avoid an increased number of chimeric, concatenated target molecules due to self-ligation, the adapter concentration in the reaction may be increased up to fourfold, or the template concentration decreased (see Note 8).
10. Note that only primers “quant_prim_F1” and “quant_prim_
R1” can be used, since the library is still truncated and does not yet contain the “quant_prim_F2” and “quant_prim_R2”
priming sites.
11. At this stage, “quant_primer” and “Amp_primer” pairs can be used for quantifi cation. For the 454 platform, “Amp_prim_F”
and “Amp_prim_R” are identical in their sequence to the
emPCR primers provided by Roche and should be preferred at this point, since they will best mimic the downstream emulsion PCR reaction and usually give more consistent and reproducible quantifi cation results.
12. On the gel, the fi nal libraries might appear to comprise DNA fragments that are longer in size than expected. This is most likely the result of heteroduplexes (library molecules that consist of two noncomplementary strands, originating from different template molecules which only hybridize in the fl anking adapter regions) migrating slower than perfectly double-stranded molecules in the gel.
170
M. Knapp
et al.
Acknowledgments
This work was supported by the Max Planck Society, the German Research Foundation (DFG), the Allan Wilson Centre for
Molecular Ecology and Evolution, the University of Otago and Pennsylvania State University.
References
1. Sanger F, Nicklen S, Coulson AR (1977) DNA
8. Meyerhans A, Vartanian JP, Wain-Hobson S
sequencing with chain-terminating inhibitors.
(1990) DNA recombination during PCR.
Proc Natl Acad Sci U S A 74:5463–5467
Nucleic Acids Res 18:1687–1691
2. Knapp M, Hofreiter M (2010) Next genera—
9. Nikiforov TT, Rendle RB, Kotewicz ML,
tion sequencing of ancient DNA: require—
Rogers YH (1994) The use of phosphorothio—
ments, strategies and perspectives. Genes 1:
ate primers and exonuclease hydrolysis for the
227–243
preparation of single-stranded PCR products
3. Meyer M, Stenzel U, Hofreiter M (2008)
and their detection by solid-phase hybridiza—
Parallel tagged sequencing on the 454 plat—
tion. PCR Methods Appl 3:285–291
form. Nat Protoc 3:267–278
10. Craig DW, Pearson JV, Szelinger S et al (2008)
4. Meyer M, Kircher M (2010) Illumina sequencing
Identifi cation of genetic variants using bar—
library preparation for highly multiplexed
coded multiplexed sequencing. Nat Methods
target capture and sequencing. Cold Spring
5:887–893
Harb Protoc. doi: 10.1101/pdb.prot5448
11. Meyer M, Briggs AW, Maricic T et al (2008)
5. Margulies M, Egholm M, Altman WE et al
From micrograms to picograms: quantitative
(2005) Genome sequencing in microfabricated
PCR reduces the material demands of high—
high-density picolitre reactors. Nature
throughput sequencing. Nucleic Acids Res 36:e5
437:376–380
12. Heyn P, Stenzel U, Briggs AW et al (2010)
6. Maricic T, Pääbo S (2009) Optimization of
Road blocks on paleogenomes—polymerase
454 sequencing library preparation from small
extension profi ling reveals the frequency of
amounts of DNA permits sequence determina—
blocking lesions in ancient DNA. Nucleic Acids
tion of both DNA strands. Biotechniques
Res 38:e161
46:51–57
13. Stiller M, Knapp M, Stenzel U et al (2009)
7. Briggs AW, Good JM, Green RE et al (2009)
Direct multiplex sequencing (DMPS)-a novel
Targeted retrieval and analysis of fi ve Nean dertal
method for targeted high-throughput sequenc—
mtDNA genomes. Science 325:318–321
ing of ancient and highly degraded DNA.
Genome Res 19:1843–1848
Case Study: Targeted high-Throughput Sequencing
of Mitochondrial Genomes from Extinct Cave Bears
via Direct Multiplex PCR Sequencing (DMPS)*
Mathias Stiller
Abstract
Here I describe the use of a recently developed technique for targeted high-throughput sequencing of highly degraded DNA by direct multiplex PCR sequencing (DMPS) that was used to amplify 31 near-complete mitochondrial genomes of the extinct cave bear (
Ursus spelaeus)
. DMPS couples multiplex PCR
with the generation of barcoded sequencing libraries to be sequenced in parallel on a high-throughput sequencing platform. DMPS makes it possible to generate large amounts of targeted DNA sequence data simultaneously from multiple degraded samples such as fossil remains. In this chapter, I describe an experiment that uses DMPS with different primer sets and on both modern and ancient DNA templates.
Key words:
Cave bear ,
Ursus spelaeus
, Mitochondrial genome , Multiplex PCR , Roche 454 FLX
platform , High-throughput sequencing , Target enrichment 1. Introduction
Using traditional PCR, cloning and Sanger sequencing, and an ancient specimen with average DNA preservation, many amplifi cations would be necessary to obtain the complete mitochondrial genome sequence from a single individual
( 1 )
. The process would necessarily consume a large amount of irreplaceable tissue (e.g., bone) in order to provide suffi cient amounts of DNA extract for all the amplifi cation reactions. A two-step multiplex PCR approach, such as that described in Chapter 17
( 13
) dramatically reduces *
Note
: In the case study presented in this chapter, I describe the amplifi cation and sequencing of whole mitochondrial genomes using a combined approach of the methods presented in Chapters 15
( 12
) , and 19
( 11
) . I discuss specifi c challenges associated with using this method to amplify and sequence modern and ancient DNA templates. For more information, see the original publication of the scientifi c results in Stiller
et al.
(2009)
( 4
) .
Beth Shapiro and Michael Hofreiter (eds.),
Ancient DNA: Methods and Protocols
, Methods in Molecular Biology, vol. 840, DOI 10.1007/978-1-61779-516-9_20, © Springer Science+Business Media, LLC 2012
171
172
M. Stiller
both the amount of time and extract required to produce these data. Tagging pr
otocols ( 2, 3
) can further simplify the process, where all second-step multiplex PCR products are barcoded, pooled, and converted into a sequencing library, and these libraries sequenced collectively using a high-throughput sequencing platform. This process can be even further simplifi ed by directly coupling the multiplex PCR and the barcoding and library preparation steps
( 4
) . Using this approach, called direct multiplex PCR sequencing (DMPS), barcoded sequencing adapters are immediately added to the fi rst-step multiplex PCR reaction, and all of the second-step reactions can be omitted. DMPS enables long, continuous
sequences to be obtained rapidly from multiple individuals.
I describe the use of DMPS to generate 31 near-complete
mitochondrial genomes of cave bears (
U. spelaeus
). Until their
extinction about 25,000 years ago, cave bears were one of the most abundant mammalian species in Europe and Asia
( 5 )
. Analyses of their remains have revealed a large amount of morphological and genetic diversity that has been loosely divided into three major lineages:
U. spelaeus
,
U. ingressus
, and
U. deningeri kudarensis
( 6– 8 )
.
Because of the challenges associated with extracting and amplifying ancient DNA, phylogenies were based on only short (ca 285 base pair (bp)) fragments of the mitochondrial control region, and were not well r
esolved ( 6– 8
) . The mitochondrial genomes generated using DMPS were used to resolve the phylogenetic relationships among the major cave bear lineages.
2. Materials
and Methods
As part of a previously published project
( 4 )
, ancient DNA (aDNA) was extracted from 110 cave bear bone or tooth specimens representing most of the species’ geographical range using a silica-based
extraction method ( 9
) . To evaluate the DNA preservation, PCR
amplifi cation of a 175 bp fragment of the mitochondrial control region was attempted from all extracts using the primers
2620F (5
¢ -GCCCCATGCATATAAGCATG-3 ¢ ) and 2558R
(5 ¢ -GGAGCGAGAGGTACACGT-3 ¢ ). Based on these results, we selected specimens that were suffi ciently well preserved for further processing.
We used Multiplex PCR to amplify the whole mitochondrial
genome of the well-preserved specimens using two, nonoverlapping sets of primers. Each of the two primer sets consisted of 64
primer pairs and targeted fragments of between 150 and 180 bp in length. We designed all primers using the online tool primer3
( http://frodo.wi.mit.edu/primer3/ ). Because of the large number of combinations of primers and possibilities for negative interaction between them, it would have been a signifi cant challenge to create and follow a robust optimization strategy. Therefore, we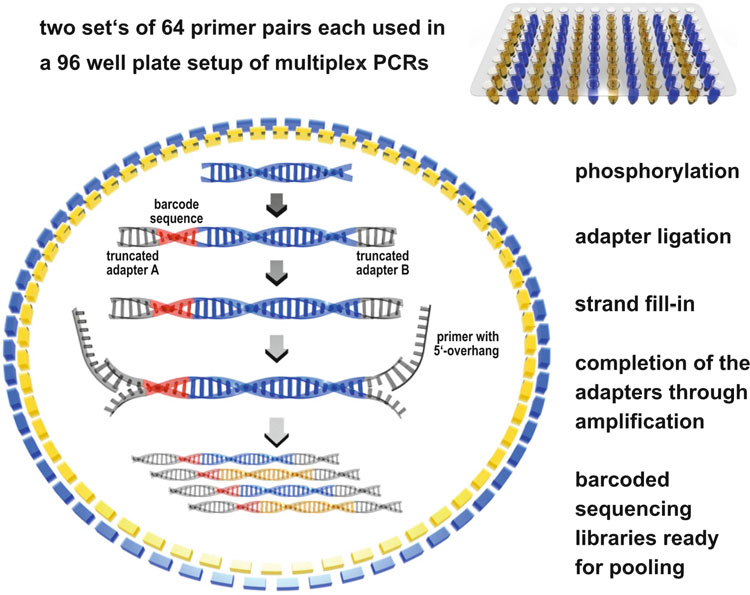
20 Case Study: Targeted high-Throughput Sequencing…
173
took no particular care to avoid primer dimer formation between primers of different fragments in each set. We then used each primer set in standard multiplex PCRs containing 2 U AmpliTaq Gold DNA polymerase, 1× AmpliTaq Gold buffer, 2.5 mM MgCl , 2
250 m M of each dNTP, 0.8 mg/mL BSA, and 150 nM of each
primer. The reactions were cycled with an activation step of 12 min at 95°C, followed by 20 cycles of denaturation at 94°C for 30 s, annealing at 53°C for 30 s, and elongation at 72°C for 30 s, with a fi nal extension step at 72°C for 10 min.