Pediatric Examination and Board Review (115 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

3.
(D)
Severe combined immunodeficiency best explains the symptoms and laboratory findings of this vignette. Although congenital HIV can present in this way, most HIV-positive children have a positive DNA PCR by 2-3 months. Patients with Wiskott-Aldrich syndrome present with recurrent pneumonia, thrombocytopenia, and eczema, the last of which this patient does not have. DiGeorge syndrome normally has dysmorphisms, hypocalcemia, and the patient is usually only mildly lymphopenic. The laboratory results do not support chronic mucocutaneous candidiasis.
4.
(E)
SCID is a heterogenous group of disorders that arise from an abnormality in the function of T cells and B cells. All of the answers are distinct genetic types of SCID, and for diagnosis, an attempt at identifying the specific genetic defect must be made; the molecular diagnosis is unknown in 14% of cases. All genetic defects leading to SCID are autosomal recessive except for one, IL2RG, which is X-linked.
5.
(B)
Although both HLA-identical and haploidentical donor stem cell transplantation are used in SCID, HLA-identical has the lowest risk of graft-versushost disease (GVHD) and a more favorable prognosis. Haploidentical donor stem cell transplantation does not have as high a success rate, but is used if necessary. IVIG is used in B-cell disorders. Although there is a promising future in gene therapy for some forms of SCID, it is currently not an approved approach to care. Without a stem cell transplantation, most patients with SCID will die before their first birthday; with transplantation before 3.5 months, the chance of survival increases to 97%.
6.
(E)
Lymphocytes are represented by CD3, CD4, and CD8 markers among others. Natural killer cells are represented by CD16, CD56, and CD57 markers. Monocytes are represented by CD14. B cells are represented by CD19 and CD20.
7.
(C)
IgA deficiency occurs in about 1 in 333 people; 1 in 700 U.S. whites are estimated to have IgA deficiency. Most are asymptomatic.
8.
(B)
The described patient has Chediak-Higashi, a disorder caused by mutation in the lysosomal transport protein. Affected individuals have recurrent skin and respiratory pyogenic infections, partial oculocutaneous albinism, and neurologic disturbances (neuropathy, photophobia, seizures). Patients who survive the infections eventually enter the “accelerated phase” of lymphocytic infiltration of most organ systems, leading to death. Laboratory findings include mild decrease in neutrophil counts with giant cytoplasmic granules.
9.
(D)
Invasive and pulmonary aspergillosis are commonly seen in several immunodeficiencies, with
Aspergillus nidulans
representing a species particularly virulent in chronic granulomatous disease. Kostmann syndrome, or congenital neutropenia, would have presented by now with recurrent infections and fever. Wiskott-Aldrich is an X-linked disorder that involves eczema, thrombocytopenia, and immunodeficiency. Common variable immunodeficiency, a B-cell disorder, would have presented after 6 months of age with encapsulated organism infections. Although LAD is also a disorder of phagocytes, it is not as commonly associated with aspergillosis.
10.
(E)
Patients with chronic granulomatous disease are susceptible to catalase-producing organisms. These organisms include, among others,
Staphylococcus
,
Burkholderia
,
Aspergillus
,
Serratia
, and
Klebsiella
species.
S pneumoniae
, however, is catalase negative.
11.
(C)
Chronic granulomatous disease results from a genetic defect in one component of the NADPHoxidase system in phagocytes (see
Figure 70-1
). These defects limit the enzyme activity that allows phagocytes to produce superoxide, which forms other reactive oxidants. This leads to an inability to defend against catalase-producing organisms.
12.
(C)
The initial screening test for CGD is the nitroblue tetrazolium (NBT) test. In this study, normal neutrophils produce superoxide that is reduced by NBT and create a dark blue coloration to cells. When the superoxide is absent or low, an abnormally low number of cells are altered to blue color. This test can then be confirmed by other more quantitative tests, such as a cytochrome
c
reduction assay.
13.
(E)
Although all of the above are possible treatments for CGD, gene therapy is currently only theoretical and studies are ongoing to test their efficacy and stability.
14.
(D)
Cutaneous delayed-type hypersensitivity testing is an in vivo test for cellular immunity. A positive test (>5-mm response in all subjects) reflects intact cellular immunity. The test uses intradermal injections of tetanus toxoid, mumps antigen, and
Candida
antigen. All negative tests should be either repeated or followed by flow cytometry and in vitro assays of T-cell function; age younger than 1 year and concurrent infection can limit the reliability of results.
15.
(D)
The mainstay of therapy for humoral deficiencies is IVIG replacement therapy.
16.
(A)
The incubation of fractionated immune globulin during the manufacturing of IVIG results in the inactivation of enveloped viruses (eg, HIV, hepatitis B, hepatitis C, herpes viruses). It does not, however, inactivate nonenveloped viruses, which include parvovirus and hepatitis A.
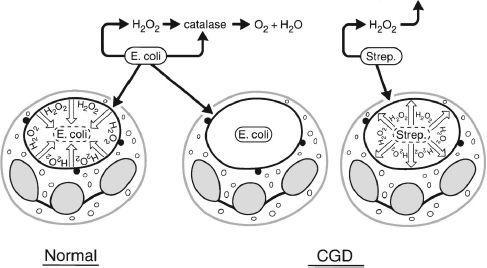
FIGURE 70-1.
Pathogenesis of chronic granulomatous disease. Schematic diagram of the manner in which the metabolic deficiency of the CGD neutrophil predisposes the host to infection. Normal neutrophils accumulate hydrogen peroxide in the phagosome containing ingested
Escherichia coli
. Myeloperoxidase is delivered to the phagosome by degranulation, as indicated by the closed circles. In this setting, hydrogen peroxide acts as a substrate for myeloperoxidase to oxidize halide to hypochlorous acid and chloramines, which kill the microbes. The quantity of hydrogen peroxide produced by normal neutrophils is sufficient to exceed the capacity of catalase, a hydrogen peroxide-catabolizing enzyme of many aerobic mieroorganisms, including most gram negative enteric bacteria,
S aureus, C albicans
, and
Aspergillus
species. When organisms such as
E coli
gain entry into the CGD neutrophils, they are not exposed to hydrogen peroxide because the neutrophils do not produce it, and the hydrogen peroxide generated by microbes themselves is destroyed by their own catalase. When CGD neutrophils ingest streptococci or pneumococci, the organisms generate enough hydrogen peroxide to result in a microbicidal effect. On the other hand, as indicated in the middle figure, catalase-positive microbes, such as
E coli
, can survive within the phagosome of the CGD neutrophil. (Reproduced, with permission, from Lichtman MA, Beutler E, Kipps TJ, et al. Williams Hematology, 7th ed. New York: McGraw-Hill; 2006: Fig 66-8.)
17.
(B)
IVIG is given every 3-4 weeks, based on its half-life, which is 21-28 days.
18.
(C)
Anaphylaxis may occur in patients with selective IgA deficiency as a result of anti-IgA antibodies reacting with trace IgA in the IVIG. For this reason, IgA-deficient patients should receive IVIG with lower IgA content.
 S
S
UGGESTED
R
EADING
Bonilla FA, Geha RS. Primary immunodeficiency diseases.
J Allergy Clin Immunol.
2003;111(2 suppl):S571-S578.
Buckley RH. Primary cellular immunodeficiencies.
J Allergy Clin Immunol.
2002;109:747-757.
Schubal SJ. Treatment of antibody deficiency syndromes.
Pediatr Rev.
2000;21:358-359.
Winkelstein JA, Marino MC, Johnston RB Jr, et al. Chronic granulomatous disease. Report on a national registry of 368 patients.
Medicine (Baltimore).
2000;79:155-169.
Chapter 8
ADOLESCENT MEDICINE
CASE 71: A 14-YEAR-OLD GIRL WHO NEEDS A SCHOOL PHYSICAL
A 14-year-old comes into the clinic for a school physical examination. She will be entering high school in the fall. She has always been healthy. Her mother reports no new problems since the last visit to her previous pediatrician 2 years earlier. She mentions that her daughter has been showing a growing interest in boys lately but has no behavioral concerns. Menses started a year ago and have been irregular. The patient wonders whether a pelvic examination will be needed on this visit. She is an excellent student and has a “good group of friends.” She is dating but denies being sexually active; she confides that many of her friends smoke cigarettes and that she has tried cigarettes in the past but did not like them. She has never used alcohol, marijuana, or other drugs. Her review of systems is negative. She lives with her mother and 2 siblings. Her father, who had type 2 diabetes mellitus, died 3 years ago of a myocardial infarction at age 47. The family history is otherwise noncontributory.
The physical examination is normal except for mild acne. She is 5'2", weighs 136 lb, and her body mass index (BMI) is 25 (91%)
SELECT THE ONE BEST ANSWER
1.
Which of the following options would you choose for conducting a physical examination in an adolescent patient?
(A) always have a parent in attendance during the physical examination