Pediatric Examination and Board Review (230 page)
Read Pediatric Examination and Board Review Online
Authors: Robert Daum,Jason Canel

(A) it is transmitted as an autosomal recessive trait
(B) little is known regarding the molecular genetics of this disorder
(C) the disorder demonstrates an X-linked mode of inheritance
(D) the disorder is the result of triplet repeat expansion
(E) the disorder is a result of mutations of mitochondrial DNA
12.
Gower sign indicates which of the following?
(A) proximal muscle weakness
(B) distal muscle weakness
(C) hypotonia
(D) high riding scapulae and facial weakness
(E) none of the above
13.
Children with congenital muscular dystrophy are deficient in which of the following proteins?
(A) dystrophin
(B) alpha-sarcoglycan
(C) F-actin
(D) dysferlin
(E) merosin
14.
You are called to the intensive care nursery to evaluate a 4-day-old male infant who is reportedly “floppy.” The infant was born at full term with Apgars of 5 and 7. Shortly after birth, he developed respiratory distress requiring mechanical ventilation. A prenatal ultrasound revealed polyhydramnios. On review of his family history, you learn that a previous child died at 36 hours of life. His maternal aunt also had a male child who died shortly after birth. On physical examination, the cranial nerves are intact with the exception of slight ptosis and a weak gag. There are no tongue fasciculations. The infant has decreased tone and absent deep tendon reflexes. There is no arthrogryposis (nonprogressive contracture of the joints). This infant most likely has which of the following disorders?
(A) nemaline myopathy
(B) central core disease
(C) spinal muscular atrophy type I
(D) centronuclear (myotubular) myopathy
(E) congenital muscular dystrophy
15.
Tongue fasciculations are most commonly seen with which of the following disorders?
(A) spinal muscular atrophy
(B) DMD muscular dystrophy
(C) nemaline myopathy
(D) centronuclear myopathy
(E) none of the above
MATCH EACH OF THE FOLLOWING DISEASES WITH ITS CORRESPONDING DEFECT OR DEFICIENCY
16. | (A) myophosphorylase (B) acid maltase |
17. | (C) tRNA Lys gene |
18. | (D) phosphorylase B kinase |
(E) lactate dehydrogenase |
ANSWERS
1.
(E)
In many cases the clinician can make the diagnosis of myasthenia gravis based on the history and physical examination (see
Figure 134-1
). The most specific test for myasthenia gravis is the detection of acetylcholine receptor antibodies in the patient’s serum. However, it should be remembered that antibodies are not detectable in all patients with myasthenia gravis. In fact, children with myasthenia gravis often do not have these antibodies. Single-fiber electromyography (EMG), nerve conduction studies with repetitive stimulation, antibody testing, and an edrophonium test can confirm the diagnosis. Edrophonium testing involves the administration of intravenous drug to patients with suspected myasthenia gravis. The drug is a cholinesterase inhibitor. A positive test consists of clinical improvement following administration of the drug, such as resolution of dysarthria or improved ocular motility. Neuroimaging, lumbar puncture, and an EEG are all likely to be normal in this patient given the clinical history.
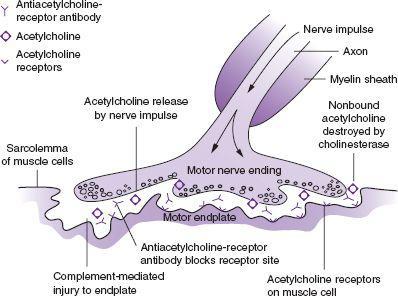
FIGURE 134-1
.
Pathogenesis of myasthenia gravis. Acetylcholine released at the nerve ending by the nerve impulse normally binds with acetylcholine receptors. This evokes the action potential in the muscle. In myasthenia gravis, anti-acetylcholine receptor antibody evokes immune-mediated destruction of the end plate. (Reproduced, with permission, from McPhee SJ, Papadakis MA. Current Medical Diagnosis and Treatment 2010, 49th ed. New York: McGraw-Hill; 2010.)
2.
(D)
Nearly a quarter of patients with myasthenia gravis develop the disease during childhood and adolescence. The condition is characterized by muscles weakness and, importantly, fatigability. Muscles of the eye, oropharynx, and extremities can be involved. In addition, respiration can be affected. Exertion and stress can bring on the symptoms. There are different categories of myasthenia gravis. This patient presents with juvenile myasthenia gravis in which onset is usually after the age of 10 years. It is more common in girls. Typically, most patients present with ocular findings, such as ptosis. However, extremity weakness can be the initial presenting sign and usually principally involves the proximal muscle groups. Neonatal myasthenia gravis is a transient condition resulting from the placental transfer of acetylcholine receptor antibodies to the fetus. Congenital myasthenic syndromes are a collection of rare diseases that are not autoimmune related. In these patients, antibodies are not found and immunosuppressive therapy is ineffective. Most patients develop symptoms in the first few years of life. Symptoms include limb weakness, ocular findings, respiratory dysfunction, and feeding difficulties. Botulism can result in dysarthria, dysphagia, ptosis, and blurred vision. In infantile botulism, hypotonia is commonly observed. This condition is caused by a toxin produced by
Clostridium botulinum
, which acts on presynaptic nerve terminals to block calcium-dependent acetylcholine release.
3.
(A)
As stated above, myasthenia gravis is more common in girls than in boys. Ptosis is the most common presenting clinical complaint with a third of patients presenting with a unilateral onset. Myasthenia gravis is frequently associated with rheumatoid arthritis, thyroid disease, diabetes, and systemic lupus erythematosus. Placental transfer of anti-AchR antibodies results in transient symptoms in the neonate.
4.
(C)
Although some debate exists, it is generally accepted that most pubertal patients with juvenile myasthenia gravis will benefit from a thymectomy with or without thymoma. The presence of a thymoma is an absolute indication for thymectomy in patients with juvenile myasthenia gravis. As a result, it is imperative that all newly diagnosed patients with myasthenia gravis undergo a chest CT to evaluate for thymoma.
5.
(E)
The main treatment of myasthenia gravis is the use of acetylcholinesterase inhibitors, such as pyridostigmine. Side effects include diarrhea, nausea, and gastrointestinal cramping. In addition, an immunosuppressive agent is usually employed, such as a corticosteroid. Azathioprine, cyclosporine, and cyclophosphamide have also been used. Azathioprine has been used when patients relapse on corticosteroids or as a steroid-sparing agent. Other forms of therapy that have been tried include plasmapheresis and the administration of intravenous immunoglobulin. Thymectomy has been considered above.
6.
(B)
This patient has developed a cholinergic crisis, with respiratory failure secondary to an overdose of acetylcholinesterase inhibitors. Overstimulation of the muscarinic and nicotinic cholinergic receptors results in nausea, abdominal cramps, excessive secretions, and bronchospasm. The term
myasthenic crisis
refers to respiratory failure that results from the patient’s disease. Treatment of the patient with cholinergic crisis involves intubation and mechanical ventilation. The patient is admitted to the intensive care unit and acetylcholinesterase inhibitors are discontinued.
7.
(E)
Patients with myasthenia gravis can have worsening of their disease should they take medications that interfere with neuromuscular transmission. Antibiotics such as sulfonamides, clindamycin, fluoroquinolones, and tetracycline can also exacerbate myasthenia gravis. Aminoglycosides are the most commonly implicated antimicrobial agents.
8.
(A)
Many patients with Duchenne muscular dystrophy (DMD) will have a small percent of dystrophinpositive “revertant fibers” which are believed to be the result of alternative splicing or secondary mutation events that restore an open reading frame. In this case, it serves as a positive control for the dystrophin staining. A subset of children with congenital muscular dystrophy lack merosin. Boys with DMD typically present with muscle weakness of the hip girdle muscles that produce a waddling gait. Later in the course, shoulder girdle muscles are affected. Smaller children may have a Gower sign (see later) when attempting to stand up from a sitting position on the floor. Calf hypertrophy is also observed. This is best characterized as a pseudohypertrophy as the apparent increase in muscle mass is caused by normal muscle being replaced by collagen and adipose tissue. Limb girdle muscular dystrophies (LGMD) represent a heterogeneous group of muscle disorders that are inherited in an autosomal dominant (LGMD1) or autosomal recessive (LGMD2) fashion. Mutations of calpain 3, alpha-sarcoglycan, and dysferlin, to name a few, result in this phenotype. This condition should be considered if dystrophin mutations have been excluded. Scapuloperoneal muscular dystrophy is a dominantly inherited condition characterized by leg weakness, foot drop (peroneal and anterior tibial muscle wasting), proximal arm weakness, and scapular winging. The calf muscles are usually unaffected. Myotonic dystrophy is inherited in an autosomal dominant fashion and caused by a trinucleotide repeat expansion mutation of a protein kinase gene. Clinically, patients with myotonic dystrophy have a high degree of variability. While some patients can be asymptomatic, others can be profoundly affected by the disease. For example, neonates may have severe hypotonia, swallowing difficulties, and respiratory distress requiring ventilatory support. Classically, patients have elongated facies, myotonia, facial diplegia, ptosis, tented upper lip, and wasting of the temporalis muscles.
9.
(C)
DMD is transmitted as an X-linked recessive disorder with mutations occurring on the short arm of the chromosome (Xp21). Most males with DMD have an absence of dystrophin.
10.
(C)
Becker muscular dystrophy is transmitted as an X-linked recessive disorder with mutations occurring on the short arm of the chromosome (Xp21). In contrast to DMD, dystrophin is present in reduced amounts.
11.
(D)
Myotonic dystrophy is inherited as an autosomal dominant trait (chromosome 19). Expansion of the triplet repeat (CTG) results in decreased expression of a protein kinase gene. This disorder demonstrates the phenomenon of anticipation; the severity of the disease progresses in subsequent generations.
12.
(A)
Gower sign occurs when a child uses all 4 limbs to push himself up off the floor from a sitting position. The child then braces his hands on his knees and thighs to push himself into an upright position. The maneuver is necessary because patients with DMD have hip muscle weakness, primarily of the gluteus maximus.
13.
(E)
Congenital muscular dystrophies (CMD) refer to a group of disorders characterized by proximal muscle weakness and hypotonia presenting in early infancy. The diseases can occur with or without major brain malformations. A portion of children with CMD are deficient in merosin (alpha
2
chain of laminin). Patients with dysferlin and sarcoglycan mutations have limb girdle muscular dystrophies types 2B and 2D, respectively.