Junk DNA: A Journey Through the Dark Matter of the Genome (25 page)
Read Junk DNA: A Journey Through the Dark Matter of the Genome Online
Authors: Nessa Carey

This is important to our cells, because most of our genes are all over the place.
a
2
By this we mean that genes are scattered around on our 23 pairs of chromosomes in a fairly nonsensical way. As we have already seen, the genes that code for the proteins required to
make haemoglobin are brought together by changes in the three-dimensional arrangements of chromosomes. This compensates for the fact that they aren’t arranged next to each other in a nice, neat way. If we look at how most of our genes are distributed, they are like the donations to a jumble sale or charity shop before they’ve been organised sensibly.
This can mean that our cells contain a gene that codes for a protein required in the foetal liver next to a gene for a protein expressed in the adult skin. There’s a huge number of such situations and this creates potential difficulties. It means that our cells require barriers between different elements to maintain different patterns of gene expression. The control needs to be relevant to a specific cell type, and to the particular developmental stage. We don’t want teeth genes expressed in our eyes or heart genes expressed in our bladders.
We know that epigenetic modifications influence gene expression. If we take the brain as an example, there are some genes that are never expressed in neuronal cells. For instance, the protein keratin is used in hair and nails, but isn’t used by our adult grey matter. In brain cells, the keratin gene is switched off and it’s kept in an inactive state by a particular pattern of epigenetic modifications. But as we’ve already seen, epigenetic modifications are blind to DNA sequence. What’s to stop these repressive modifications from creeping along from the keratin gene and starting to switch off other genes as well?
This is particularly a problem because epigenetic modifications are often self-sustaining. Let’s take the case of modifications that are involved in repressing gene expression. These modifications attract other proteins that reinforce the initial change, making it even harder to reactivate gene expression. These in turn can attract proteins that continue to add repressing epigenetic modifications, to prevent escape from inactivation. But we can imagine that the borders of the repression are quite vague, because the epigenetic
machinery doesn’t recognise specific DNA sequences. So, at the periphery of the repressed regions, the epigenetic modifications could spread out.
Halting the spread
Our cells have evolved a remarkable way to prevent this. Just as fire crews will cut down stands of trees or blow up buildings to create a gap in the path of an inferno, our genome removes the fuel for the epigenetic machinery. Junk DNA that acts as an insulator between repressed and active regions of the genome loses its histone proteins. No histone proteins means no epigenetic histone modifications. No modifications means no spreading of epigenetic activity. This stops repressive modifications creeping into active genes and also prevents the opposite effect. This is shown in Figure 13.1.
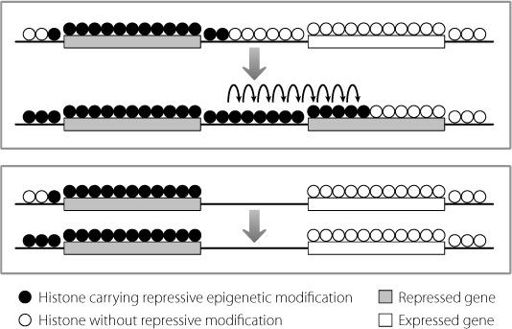
Figure 13.1
In the upper panel, repressive modification patterns spread from one gene to the next. In the lower panel, the lack of histones in the insulator regions between two genes prevents the spread of the repressive epigenetic modifications, and stops the right-hand gene from being abnormally silenced.
But because different cells need to insulate different regions (we do, after all, want keratin expressed in the cells that create hair) we can deduce that DNA sequence alone isn’t enough to create an insulator. Instead, these are generated by complex, situationally dependent interactions between the genome and the combinations of proteins expressed in a cell at any one time.
One of the most important of these proteins is a ubiquitously expressed one that we can refer to as 11-FINGERS.
b
It’s a large, highly conserved protein with a characteristic structure. The way that it folds in three dimensions means that there are eleven finger-like projections that stick out from the protein. Each of these eleven fingers can recognise a defined DNA sequence, but not each finger recognises the same sequence.
Imagine an eleven-fingered pianist wearing gloves where the wool on each digit is one of four colours. Combine this with a piano where each key is also one of the same four colours, assigned randomly between the keys. The rules are that the pianist can play any notes she likes, but must always hit between two and eleven notes simultaneously, and the colours on the fingers and keys must match. We can start to see that there are an awful lot of possible combinations. And to understand the extent of the different options, now imagine that the piano has thousands of keys.
The 11-FINGER protein is able to bind to lots of different genomic sequences in a similar way. It can bind to tens of thousands of sites in human cells. In addition to binding itself to DNA, 11-FINGER also binds other proteins. We can again invoke our abnormally digited piano player to visualise this. Imagine there is Velcro on the backs of the gloves, which can bind fuzzy balls of fluff. The coloured fingers of the gloves hit the piano keys, the backs of the gloves get covered in fluffy fabric balls.
So it is for 11-FINGER. The finger-like projections bind to
DNA, the other surfaces of the protein bind other proteins. The precise binding partners will depend on the complement of proteins being expressed in a cell. One of the proteins can alter the coiling of DNA, which can be important for controlling gene expression.
3
Another is a protein that deposits specific epigenetic modifications.
4
In some regions the types of genomic interlopers we met in Chapter 4 serve as insulators, preventing the spread of activating or repressive epigenetic modifications from one region to another.
5
Some tRNA genes can act as insulators. They can stop expression of one gene driving inappropriate expression of a neighbouring gene. This is an additional benefit of having lots of tRNA genes, which demonstrates the economical way with which evolution has made the most of raw material.
The way this works is shown in Figure 13.2. A classical protein-coding gene is coated with epigenetic modifications that promote its expression. The enzyme that binds to this gene and copies it into RNA (which will ultimately be processed to form mature messenger RNA) can be a bit of a runaway train: once it starts copying it tends to keep going. If there is another protein-coding gene nearby, the enzyme could keep going and copy this as well. But if there are two or more tRNA genes in between, this won’t happen. tRNA genes are switched on pretty much all the time, because they are involved in the creation of all proteins. There is an enzyme that copies tRNA genes to create tRNA molecules from the DNA template. But this is different from the enzyme that carries out a similar job to generate messenger RNA molecules from classical protein-coding genes. The enzyme that creates the tRNA molecules acts like a big burly bouncer, stopping the other enzyme from getting through the door to the next gene. Because the enzyme that copies tRNA genes can’t bind to classical protein-coding genes, this keeps the overall gene expression in this region under tight spatial control.
6
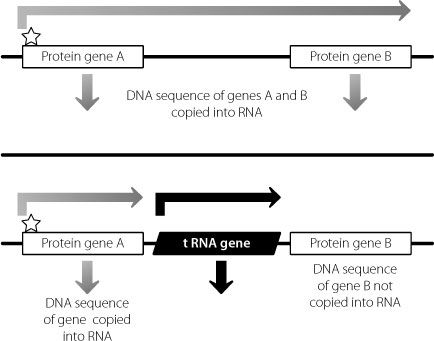
Figure 13.2
The enzyme that copies DNA into messenger RNA from protein-coding genes binds at the star at the start of gene A. If nothing stops it, the enzyme could keep on copying until it has also copied protein-coding gene B into messenger RNA, perhaps inappropriately. tRNA genes are copied from DNA into functional tRNA molecules by a different enzyme. This blocks the progress of the enzyme creating messenger RNA from gene A, and prevents inappropriate use of gene B.
Because there has been such an emphasis in biology on the dividends from the development of DNA sequencing technologies, it’s always tempting to think that most of the big conceptual breakthroughs arise from high-end molecular approaches. But the reality is that basic human biology and logical thought actually take us a long way.
Why XX is different from XXX
In Chapter 7 we saw that female mammals always inactivate one X chromosome in their cells, to ensure that they have the same levels of X chromosome gene expression as male cells. Our cells are able to count. If a female cell contains three X chromosomes, Protein gene B DNA sequence of genes A and B copied into RNA DNA sequence of gene copied into RNA
it will switch off two of them. Conversely, if there is only one X chromosome, the cell leaves this switched on.
This leads us to a pretty obvious prediction. It doesn’t matter how many X chromosomes a cell contains, because X inactivation will always ensure that only one is functionally active. Therefore, as long as a person contains at least one X chromosome in each cell, they will be completely normal and healthy.
The problem is, this isn’t true. Women with only one X chromosome, or with three X chromosomes, do have detectable symptoms. So do men who have two X chromosomes in addition to their Y. One explanation could be that maybe X inactivation isn’t working well in these people, but that doesn’t seem to be the case. X inactivation is a very robust system. It’s unlikely to work perfectly every single time – nothing else in biology does. But random inadequacies in the system wouldn’t explain why all women with just one X chromosome present with very similar clinical symptoms.
Women with just one X chromosome are shorter than average, and have underdeveloped ovaries.
7
Women with three X chromosomes are taller than average and at increased risk of learning disabilities and developmental delay as children.
8
Males with two X chromosomes (plus a Y of course) are taller than average, and may have relatively small testicles, leading to problems caused by low production of the male hormone, testosterone. They are also at increased risk of learning disabilities.
9
Although potentially distressing for the patients and their families, the symptoms are milder than we see for patients with abnormal numbers of autosomal chromosomes (remember Down’s, Edward’s and Patau sydromes –
see pages 76–7
). That’s because although the X chromosome is large, most of the genes on it are appropriately inactivated, no matter how many copies of this chromosome are present. But there are some that aren’t.
To understand what is happening, we need to think back to what happens when eggs or sperm are created. At a certain stage,
the chromosomes line up in pairs and then one of each pair is pulled to opposite ends of the cell. The cell divides and its daughter cells contain one of each pair. In a female cell this is easy to visualise. The two X chromosomes pair up and then can be separated, in exactly the same way as any other pair of chromosomes from number 1 to number 22. But when males are creating sperm, there is a problem. Males contain one large X chromosome and one tiny Y chromosome. These are very different from each other. Yet somehow, during the creation of sperm, the X and the Y must find each other and pair up, despite being so different.
The reason they can do this is because there is a small region at the ends of the X and Y chromosomes where they are very similar to each other. This allows them to recognise each other and to associate during cell division, holding hands until they need to move to opposite ends of the dance floor.
These stretches are known as pseudoautosomal regions. They contain protein-coding genes, and they are protected from silencing during X inactivation. The genes in the pseudoautosomal region are treated very differently from most of the other genes on the X chromosome. This pattern of activated and inactivated genes, which leads to detectable symptoms in males and females with the wrong number of X chromosomes, was a clear sign from biology that cells contain very fundamental ways of functionally separating different blocks of DNA.