Junk DNA: A Journey Through the Dark Matter of the Genome (27 page)
Read Junk DNA: A Journey Through the Dark Matter of the Genome Online
Authors: Nessa Carey

Most gene promoters were associated with more than one of these regions, and each region was typically associated with more than one promoter. Yet again, it appears that our cells don’t use straight lines to control gene expression, they use complex networks of interacting nodes.
Some of the most striking data suggested that over 75 per cent of the genome was copied into RNA at some point in some cells.
9
This was quite remarkable. No one had ever anticipated that nearly three-quarters of the junk DNA in our cells would actually be used to make RNA. When they compared protein-coding messenger RNAs with long non-coding RNAs the researchers found a major difference in the patterns of expression. In the fifteen cell lines they studied, protein-coding messenger RNAs were much more likely to be expressed in all cell lines than the long non-coding RNAs, as shown in Figure 14.3. The conclusion they reached from this finding was that long non-coding RNAs are critically important in regulating cell fate.
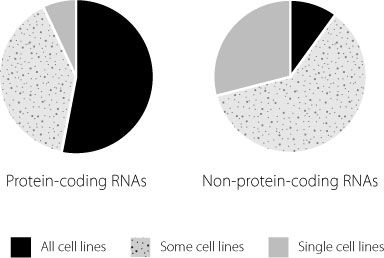
Figure 14.3
Expression of protein-coding and non-coding genes was analysed in fifteen different cell types. Protein-coding genes were much more likely to be expressed in all cell types than was the case for regions that produced non-coding RNA molecules.
Taken in their entirety, data in the various papers from the
ENCODE consortium painted a picture of a very active human genome, with extraordinarily complex patterns of cross-talk and interactivity. Essentially the junk DNA is crammed full of information and instructions. It’s worth repeating the hypothetical stage directions from the Introduction: ‘If performing
Hamlet
in Vancouver and
The Tempest
in Perth, then put the stress on the fourth syllable of this line of
Macbeth
. Unless there’s an amateur production of
Richard III
in Mombasa and it’s raining in Quito.’
10
This all sounds very exciting, so why was there a considerable degree of scepticism about how significant these data are? Part of the reason is that the ENCODE papers made such large claims about the genome, particularly the statement that 80 per cent of the human genome is functional. The problem is that some of these claims are based on indirect measures of function. This was especially true for the studies where function was inferred either from the presence of epigenetic modifications or from other physical characteristics of the DNA and its associated proteins.
Potential versus actual
The sceptics argue that at best these data indicate a
potential
for a region to be functional, and that this is too vague to be useful. An analogy might help here. Imagine an enormous mansion, but one where the owners have fallen on hard times and the power has been disconnected. Think Downton Abbey in the hands of a very bad gambler. There could be 200 rooms and five light switches in each room. Each switch could potentially turn on a bulb, but it may be that some of the switches were never wired up (aristocrats are not known for their electrical talents), or the associated bulb is broken. Just because the switches are on the walls, and can be flicked between the on and off positions, it doesn’t actually tell us that they will really make a difference to the level of light in the room.
The same situation may take place in our genomes. There may be regions that carry epigenetic modifications, or have specific physical characteristics. But this isn’t enough to demonstrate that they are functional. These characteristics may simply have developed as a side effect of something that happened nearby.
Look at any photo of Jackson Pollock creating one of his abstract expressionist masterpieces.
11
It’s a pretty safe bet that the floor of his studio got spattered with paint as he created his canvases. But that doesn’t mean that the paint spatters on the floor were part of the painting, or that the artist endowed them with any meaning. They were just an inevitable and unimportant by-product of the main event. The same may be true of physical changes to our DNA.
Another reason why some observers have been sceptical about the claims made in ENCODE is based on the sensitivity of the techniques used. The researchers were able to use methods that were far more sensitive than those employed when we first started exploring the genome. This allowed them to detect very small amounts of RNA. Critics are afraid that the techniques are too sensitive and that we are detecting background noise from the genome. If you are old enough to remember audio tapes, think back to what happened if you turned the volume on your tape deck really high. You usually heard a hissing noise behind the music. But this wasn’t a sound that had been laid down as part of the music, it was just an inevitable by-product of the technical limitations of the recording medium. Critics of ENCODE believe that a similar phenomenon may also occur in cells, with a small degree of leaky expression of random RNA molecules from active regions of the genome. In this model, the cell isn’t actively switching on these RNAs, they are just being copied accidentally and at very low levels because there is a lot of copying happening in the neighbourhood. A rising tide lifts all boats, but it will also raise any old bits of wood and abandoned plastic bottles that happen to be in the water as well.
This seems like a quite significant problem when we realise that in some cases the researchers detected less than one molecule of a particular RNA per cell. It isn’t possible for a cell to express somewhere between zero copies and one copy of an RNA molecule. A single cell either produces no copies of that particular RNA or it makes one or more copies. Anything else is like being ‘sort of’ pregnant. You’re either pregnant or you’re not, there’s nothing in between.
But this doesn’t actually tell us that the techniques used were too sensitive. Instead, it tells us that our techniques still aren’t sensitive enough. Our methods aren’t good enough to allow us to isolate single cells and analyse all the RNA molecules in that cell. Instead we have to rely on isolating multiple cells, analysing all the RNA molecules and then calculating how many molecules were present on average in the cells.
The problem with this is that it means we can’t distinguish between a large percentage of the cells in a sample each expressing a small amount of a specific RNA and a small percentage of the cells each expressing a large amount of the RNA. These different scenarios are shown in Figure 14.4.
The other problem we have is that we have to kill the cells in order to analyse the RNA molecules. As a consequence we only generate snapshots of the RNA expression where ideally we would want to have the equivalent of a movie so that we could see what was happening to RNA expression in real time. The problem inherent in this is demonstrated in Figure 14.5.
Ideally, of course, we should be able to test if the findings of ENCODE really stand up to scrutiny by direct experiments. But this takes us back to the problem that there were so many findings. How do we decide on candidate regions or RNA molecules to study? The additional complication is that many of the features identified in the ENCODE papers are parts of large, complex networks of interactions. Each component may have a limited effect on its own in the overall picture. After all, if you cut through one
knot in a fishing net, you won’t destroy the overall function. The hole may allow the occasional fish to escape but losing one small fish probably won’t have too much impact on your catch. Yet that doesn’t mean all the knots are unimportant. They all are important, because they work together.
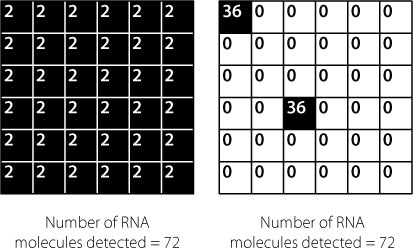
Figure 14.4
Each small square represents an individual cell. The figures inside the cell indicate the number of molecules of a specific RNA molecule produced in that cell. The researcher analyses a batch of cells, due to the sensitivity limits of the detection methods available. This means that the researcher only has access to the total number of molecules in the batch and cannot distinguish between (
left
) 36 cells all containing two molecules and (
right
) two cells (out of 36) each containing 36 molecules – or any other combination that results in an overall total of 72 molecules.
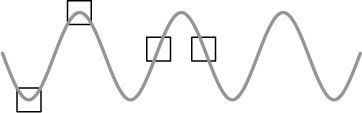
Figure 14.5
Expression of a specific RNA in a cell may follow a cyclical pattern. The boxes represent the points at which a researcher samples the cells to measure expression of that RNA molecule. The results may appear very different when comparing different batches of cells, perhaps from discrete tissues, but it may be that this simply reflects a temporal fluctuation rather than a genuine, biologically significant variation.
The evolutionary battleground
The authors of the ENCODE papers, and of the accompanying commentaries, also used the data to draw evolutionary conclusions about the human genome. Part of the reason for this lay in an apparent mismatch. If 80 per cent of the human genome has function, you would predict that there should be a significant degree of similarity between the human genome and at the very least the genomes of other mammals. The problem is that only about 5 per cent of the human genome is conserved across the mammalian class, and the conserved regions are highly biased towards the protein-coding entities.
12
In order to address this apparent inconsistency, the authors speculated that the regulatory regions have evolved very recently, and mainly in primates. Using data from a large-scale study of DNA sequence variation in different human populations, they concluded that the regulatory regions have relatively low diversity in humans, whereas the diversity is much higher in areas that have no activity at all. One of the commentaries explored this further, using the following argument. Protein-coding sequences are highly conserved in evolution because a particular protein is often used in more than one tissue or cell type. If the protein changed in sequence, the altered protein might function better in a particular tissue. But that same change might have a really damaging effect in another tissue that relies on the same protein. This acts as an evolutionary pressure that maintains protein sequence.
But regulatory RNAs, which don’t code for proteins, tend to be more tissue-specific. Therefore they are under less evolutionary pressure because only one tissue relies on a regulatory RNA, and possibly only during certain periods of life or in response to certain environmental changes. This has removed the evolutionary brakes on the regulatory RNAs and allowed us to diverge from our mammalian cousins in these regions. But across human populations, there has been pressure from evolution to maintain the optimal sequence for these regulatory RNAs.
13
Biologists tend to be a rather restrained social group when it comes to disagreements. There’s the occasional aggressive question-and-answer session at a conference but generally public pronouncements are carefully phrased. This is especially true of anything that is published, rather than said at a meeting. We all know how to read between the lines, of course, as shown in Figure 14.6 but typically, published papers are carefully phrased. That’s what made the debate that followed ENCODE particularly entertaining to the relatively disinterested observer.