Junk DNA: A Journey Through the Dark Matter of the Genome (30 page)
Read Junk DNA: A Journey Through the Dark Matter of the Genome Online
Authors: Nessa Carey

But if we look instead at schizophrenia, we find that if one twin suffers from this condition, there is only a 50 per cent chance that their identical twin is also affected. This has been calculated by analysing lots of twin pairs and working out the frequency with which both twins develop the condition. This tells us that genetics contributes about half the risk for developing schizophrenia and the other risk factors aren’t due to the genome.
Researchers can extend these studies into other family members, because we know how much genetic information family members share. For example, non-identical siblings share 50 per cent of their genetic information, as do parents and children. First cousins share only 12.5 per cent of their genomes. It’s possible to use this information to calculate the contribution of genetics to a large range of conditions from rheumatoid arthritis to diabetes, and from multiple sclerosis to Alzheimer’s disease. In these conditions, and many more, genetics and environment act together.
If we can find enough families, we can analyse their genomes to identify regions that are associated with disease. But we have to remember that the data we generate will be very different from
the simple situation we see with a purely genetic condition such as Huntington’s disease. In Huntington’s 100 per cent of the genetic contribution lies in one mutation in one protein-coding gene. But for a condition such as schizophrenia, the 50 per cent genetic contribution to the disease isn’t due to just one gene, and the same is true of most other conditions where both genetics and the environment play a role. There could be five genes each contributing 10 per cent of the risk for schizophrenia, or twenty genes each contributing 2.5 per cent of the risk. Or any other combination of which you can think. This makes it harder to identify the relevant genetic factors, and to prove that sequence changes really do influence the condition being studied.
Notwithstanding these difficulties, more than 80 diseases and traits have been mapped using these methods, generating thousands of candidate regions and variations.
h
,
14
Remarkably, nearly 90 per cent of the regions identified across these studies are in junk DNA. About half are in the regions between genes, and the other half are in the junk regions within genes.
15
Guilt by association
We have to be very careful about assuming that because we can detect a variation in DNA that is associated with disease, that this means the variation has a role in causing the disease. Sometimes we may just be looking at guilt by association. The genetic change that really contributes to the condition may be a different variation that is close by, and our candidate may just have been carried along for the ride.
An example of guilt by association would be cirrhosis of the liver. One way to assess exposure to cigarette smoke is to measure
the levels of carbon monoxide in a person’s breath. Ten years ago, if we measured the levels of this gas in the breath of non-smokers with liver disease we might find that there were higher concentrations of this gas in the airways of people with the condition than without, on average. One interpretation (although not the only one) would be that passive smoking increases the risk of liver cirrhosis. But in reality, the carbon monoxide levels are a case of guilt by association. They probably just reflect that the patient may spend a lot of time in pubs and bars, because excessive alcohol consumption is a major risk factor for developing this illness. Until the introduction of smoking bans in many cities, pubs and bars were traditionally pretty smoke-filled environments.
Even if we exclude guilt by association when analysing the contribution of a genetic variation to a human disorder, we still need to be really careful to test hypotheses about the functional consequences of our findings. Otherwise, we can be badly misled.
The variation that contributes to human pigmentation that we met earlier in this chapter actually lies in the introns, the bits of junk DNA that lie in between the protein-coding parts of a gene, which we first met in Chapter 2. The gene is very big, and the variant base pair is in the 86th stretch of junk DNA between amino acid-coding regions. But this gene itself plays no role in control of pigment levels. So we have clear precedent for accepting that variations in the junk regions in one gene may be important for effects on other genes.
Obesity is one area in which there has been a great deal of interest in identifying genetic variants linked to physical variation. Nearly 80 different regions in the human genome have been associated with obesity or with other relevant parameters such as body mass index.
16
In multiple studies, the variation showing the greatest association with obesity was a single base pair change in a candidate
protein-coding gene on chromosome 16.
i
,
17
,
18
Individuals who inherited an A on both copies of this gene tended to be about 3kg (6.6lb) heavier than individuals who inherited a T on both copies. This change was in the junk region between the first two amino acid-coding stretches of the candidate gene. The fact that this association was detected in more than one study was important as this increases our confidence that we are looking at a meaningful event.
It seemed that a consistent story was developing, because experiments in mice seemed to confirm a role for this gene in control of body weight. Mice that were genetically manipulated so that they over-expressed this gene were overweight, and developed type 2 diabetes symptoms when they ate a high-fat diet.
19
When this gene was knocked out in mice, the animals had less fat tissue and a leaner body type than control mice. Even when the knockout mice ate a lot, they burnt loads of calories, even though they weren’t particularly active.
20
This created a lot of excitement. It implied that if scientists could find a way to inhibit the activity of this gene in humans, they might be able to develop an anti-obesity drug. There was still a problem because we aren’t altogether sure what the candidate gene does in cells, and that makes it difficult to create good drugs. But at least we had a starting point. Both the human and mouse data implied that the gene coded for an important protein in obesity and metabolism. This was coupled with the reasonable assumption that the variant base pair associated with obesity affected the expression of the gene itself.
But in the immortal words of Mitch Henessey, the character played by Samuel L. Jackson in
The Long Kiss Goodnight
, ‘Assumption makes an ass out of “u” and … “umption”.’ Of course, hindsight is always 20/20 and there’s no reason to feel
condescending to the scientists who were exploring the role of the protein. It’s just that nature seems to have a way of tripping us up.
Here’s the real reason why that single base pair variation makes a difference to human physiology. There’s another protein-coding gene half a million base pairs away from the key single base pair change described above.
j
The junk region of the original gene interacts with the promoter of the second gene, altering its expression patterns. Essentially, the junk region acts as an enhancer. The effect is seen in humans, mice and fish, suggesting it is an ancient and important interaction.
The investigators looked at the expression levels of this second gene in over 150 human brain samples. There was a clear correlation between the base pair variant in the junk/enhancer region and the expression levels of the second gene. But there was no correlation between the base pair variant and the expression levels of the original candidate, the gene that actually contains the variation.
When the researchers knocked out expression of the second gene in mice they found that, compared with control animals, the mice were lean, had low adipose tissues and increased baseline metabolic rate. This was in fact the first time anyone realised that the second gene was involved in metabolism at all.
21
What we have here is a model very similar to the one we have already encountered for human pigmentation and for pancreatic agenesis. There are in fact a number of different variant base pairs in the junk region of the original obesity-associated gene. Many of these have been associated with obesity. This suggests that all of these variants probably have the same effect, i.e. they change the activity of the enhancer, and thereby alter the expression levels of the target gene, half a million base pairs away.
Of course, the mouse data suggest that the original gene, the one that contains the variations in its junk DNA, may also play
a role in obesity and metabolism. So we could ask if, in practical terms, it really matters how the single base pair changes bring about their effects. But there is a way in which this matters a lot, and that’s in the field of drug discovery.
One of the many problems in developing new drugs is that frequently some patients will respond to a drug and others won’t. This adds a lot of additional expense. It means that pharmaceutical companies have to run very large clinical trials to see if their drug works, because they have to test it in all-comers. It also means that it’s expensive to use the drug in clinical practice, because the doctor will give it to all patients with the relevant condition, but it will only work in some of them.
These days, pharmaceutical companies are all trying to create something called ‘personalised medicine’. This means that they try to develop drugs for situations where they know very early on which patients they want to treat, usually based on their genetic background. This can be very effective. It means drugs cost less money to develop, are usually licensed faster, and are only given to patients who are likely to benefit. This is an advantage for the health care providers because they aren’t wasting money treating people who won’t respond. It’s also potentially better for the patients, as all drugs have possible side effects, and there’s no point having the risk of side effects if there is very little likelihood of receiving benefit.
22
There have been real successes in this approach, most notably in drugs for breast cancer,
23
a blood cancer
24
and most recently lung cancer.
25
The critical step in developing personalised medicines is to identify a reliable biomarker. The biomarker tells you which of your potential patients should respond to your drug. Ideally, you want a situation where 100 per cent of people with the relevant biomarker will respond to the drug. The problems start if you have the right biomarker for the disease, but you link it with the wrong target. You will create a drug and then be stuck wondering why
patients who ‘should’ respond, don’t. It will be because there is a break in the circle of relationships, as shown in Figure 15.3.
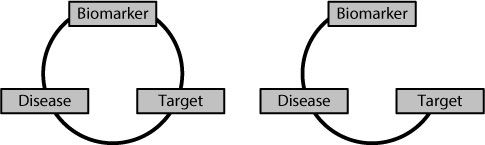
Figure 15.3
On the left-hand side there is a perfect relationship between the biomarker, target and disease. On the right-hand side there is no relationship between the target and the presence or absence of the particular biomarker. Under these conditions the biomarker is useless for predicting which patients with the disease will respond to a drug developed against the target.
The potential market for obesity drugs is, with no pun intended, huge. It’s likely some companies had already started drug discovery programmes against the original target, which they will now be either terminating, or trying to find a way to salvage. In the meantime, portion control and a bit of exercise remain our best bet.
Footnotes
a
The protein is called Sonic Hedgehog, symbol SHH. Researchers went through a phase of giving genes apparently comic names. This is now discouraged as it’s suddenly not so amusing if a genetic counsellor has to pass on a whimsical gene name to the parents of a child with a severe genetic condition.
b
The enhancer region is called ZRS and is found on the long arm of chromosome 7.
c
This transcription factor is called Six3.
d
This transcription factor is called GATA6.
e
This transcription factor is called PTF1A.
f
This variant base pair has the catchy name of rs12913932.
g
This gene is called OCA2.
h
This approach to finding disease/trait-associated genes and variants is known as GWAS – genome-wide association studies.